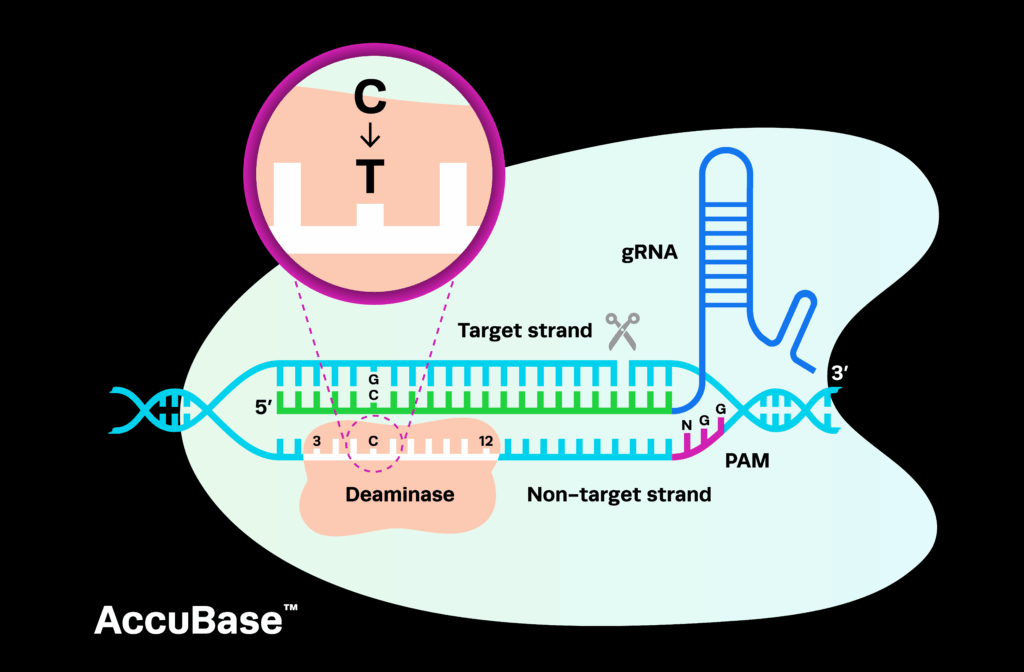
In mid-2025, a team of scientists made headlines around the world for creating and administering a bespoke CRISPR therapy to a baby with a rare genetic disorder, all in an unprecedented timeframe of six months. The patient, known as Baby KJ, is now thriving. This unique, widely publicized case heralds a new era in personalized medicine; the FDA has now used Baby KJ’s therapy as the foundation for the plausible mechanism pathway, a new regulatory framework designed for complex biologics.
In a two-page ‘sounding board’ article in the New England Journal of Medicine, Vinay Prasad, the director of the FDA’s Center for Biologics Evaluation and Research (CBER), and Marty Makary, the FDA Commissioner, discuss the basis of the plausible mechanism pathway. It’s the latest in a series of initiatives and changes at the FDA that aim to support the development of cell and gene therapies, which have struggled to fit within traditional drug development frameworks.
At face value, the plausible mechanism appears to be a fit-for-purpose regulatory framework for bespoke therapies, with a goal of cutting onerous red tape for therapeutic developers. However, it leaves a lot of room for interpretation and has raised concerns about regulatory consistency from rare disease experts. Let’s delve into the details of the paper, including why the plausible mechanism pathway was developed, the core pillars of the pathway, what types of therapies will be eligible, and how it could potentially affect CRISPR therapeutic development.
What is the Plausible Mechanism Pathway?
In their article, Prasad and Makary state that the FDA is committed to providing a pathway to market for therapies that cannot be assessed based on randomized control trials, as was the case for Baby KJ. They propose the plausible mechanism pathway as a tailor-made regulatory framework for these therapies.
The pathway was conceptualized by the FDA in collaboration with a range of stakeholders, including patients and their families, researchers, clinicians, and drug sponsors. The FDA may have had plans for a more flexible approach to regulatory approvals for rare diseases for some time, and Baby KJ’s case exemplifies best practise for such a pathway. Throughout the paper, they continually reference Baby KJ’s bespoke base editing therapy as the test case for this new approach.
Expanded Access vs. Plausible Mechanism Pathway
Baby KJ’s personalized gene therapy was assessed under a single-patient, expanded access investigational new drug (IND) application, which is typically employed to allow for patients with very limited treatment options to gain access to investigational drugs that have not yet been approved – for example, those currently being tested in clinical trials. In these cases, sponsors are not expected to use the data from those patients in their subsequent marketing application.
The distinction made in the paper is that although it was tailor-made for one individual, Baby KJ’s therapy did generate important safety and efficacy data that could be applied to the development of similar products or for the treatment of more than one mutation. Under the plausible mechanism pathway, the FDA would work toward market authorization for drug manufacturers who have demonstrated the success of bespoke therapies in multiple patients. Using platform technology, the sponsor could then potentially gain approval for similar personalized therapies.
What is unclear at this stage is how the plausible mechanism pathway will intersect with other regulatory designations and pathways, such as the accelerated approval (AA), orphan drug designation (ODD), and regenerative medicine advanced therapy (RMAT) programs. For example, the paper does not clarify if sponsors will need to file an initial IND application under the expanded access pathway and gather data on the consistency of the therapies or platform technology before they can be considered for the plausible mechanism pathway.
"The paper proposes a framework but does not clarify how it will fit with other regulatory frameworks that are similarly meant to accelerate therapeutic development. "
Lina Jamis, Director, Regulatory Affairs and Compliance at Synthego
Core Pillars of the Plausible Mechanism Pathway
According to the paper, the plausible mechanism pathway is based on five tenets that are exemplified by Baby KJ’s case. Let’s briefly explore each of these core pillars.
Identification of the specific underlying cause of disease
Rather than using a broad set of diagnostic criteria or unclear genomic associations, sponsors hoping to use the plausible mechanism pathway must identify a specific molecular/cellular abnormality that is causing the disease.
The therapy in question targets the underlying cause
Under the plausible mechanism pathway, therapies will need to address the underlying cause of the disease. The paper uses corticosteroids as an example of drugs that would not qualify for this pathway; while they treat a range of conditions, they do not target the root cause.
There is a well-characterized natural history of the disease in untreated patients
In Baby KJ’s case, there was well-established evidence that his condition would result in neurological damage if left untreated. Drugs assessed under the plausible mechanism pathway are most likely to be those that can treat patients for whom there are limited to no treatment options and/or with rapidly progressing conditions.
Confirmation that the target was successfully drugged, edited, or both
This point is potentially the most interesting and unclear aspect of the plausible mechanism pathway. The paper states that Baby KJ’s therapy was approved based on a mouse model that showed editing in 42% of liver cells, but that editing levels could not be confirmed in the real patient because a liver biopsy would place the baby at unnecessary risk. However, the authors remark that animal models are often inadequate and that the FDA will adopt non-animal models if necessary. Details here are lacking, but these alternatives may include organoids generated from patient-derived iPSCs or similar models.
Improvement in clinical outcomes or disease course
The final pillar outlined under the plausible mechanism pathway is that there must be improvements in the patient's clinical outcomes or disease progression. The authors remark that this may include using the patient’s prior clinical course as their own control.
Postmarketing Commitments Under the Plausible Mechanism Pathway
Following approval, sponsors of drugs assessed under the plausible mechanism pathway will be expected to gather evidence that supports the efficacy of the therapy, rule out off-target editing, and monitor the effects on developmental milestones in children who have been treated. The FDA and sponsor will agree on risk-benefit metrics in advance and will consider the feasibility of gathering efficacy and off-target data based on which cells or tissues are targeted.
What Types of Therapies Will Be Eligible for the Plausible Mechanism Pathway?
According to the paper, the plausible mechanism pathway will prioritize rare and fatal diseases, including progressive conditions that lead to severe childhood disabilities. Common conditions for which there is considerable unmet need will also qualify.
As an example of a well-suited case for the plausible mechanism pathway, the authors mention a disease caused by 150 mutations with the same functional consequences that would require 150 different therapies. This is another reference to Baby KJ’s condition: CPS1 deficiency can be caused by hundreds of mutations, of which KJ had one. Researchers who developed Baby KJ’s bespoke therapy can apply the same base editing technology packaged in lipid nanoparticles to target the many other mutations that cause the disease and speed up the approval process rather than starting from scratch with a new application for each mutation they target.
This could also include diseases like X-linked agammaglobulinemia, which is caused by hundreds of different mutations in the Bruton's tyrosine kinase gene. Sponsors with platform gene-editing technology could generate similar therapies targeting each mutation in very small patient populations and receive licensing approval from the FDA under the proposed pathway.
"The FDA has released at least one prior guidance on platform technologies, but has not clarified the definition of what is and is not considered a platform. For example, how much variation can there be within a platform technology for it to still be considered under the plausible mechanism pathway?"
Lina Jamis, Director, Regulatory Affairs and Compliance at Synthego
Beyond CRISPR cell and gene therapies and other complex biologics, the authors comment that there is no obstacle to other types of drugs, such as antibodies and small molecules, being assessed under the plausible mechanism pathway in due course.
Guidance Documents and Remaining Questions
The plausible mechanism pathway is certainly exciting for sponsors of CRISPR cell and gene therapies and other complex biologics, but at this stage, it remains fairly nebulous and there are no exact regulatory guidelines. Any sponsors hoping to submit a BLA for consideration under this pathway will likely need clarity on the finer details of what is required. Many questions remain regarding the use of animal models and tests to confirm levels of gene editing, and these will likely be decided on a case-by-case basis; after all, the pathway was developed based on the fact that for complex biologics, not one size fits all.
The fifth pillar of the pathway, in which the FDA may use the patient as their own control, raises key questions given recent developments in the biopharma space. For example, uniQure recently announced in a brief press release that the FDA had deemed external control group data inadequate to support the BLA submission for their AMT-130 gene therapy for Huntington’s disease. While the exact details are unclear, this disagreement with the FDA suggests that the waters are still murky for many experimental gene therapies.
Outside of the gene therapy space, a small molecule therapy for pyruvate dehydrogenase complex deficiency, an ultra-rare pediatric mitochondrial disease, was also recently rejected by the FDA, despite previously receiving orphan drug, rare pediatric disease, and priority review designations. The sponsor, Saol Therapeutics, is currently attempting to circumvent the need for a new trial by gathering additional data and performing new analyses to address the issues raised by the FDA. This case similarly highlights the difficulties sponsors face in the rare disease field, in which patient populations are exceedingly small, making it difficult to gather sufficient data.
Interestingly, Prasad and Makary also get a jump start on potential naysayers in the paper; after stating that critics may suggest the FDA’s current frameworks are sufficient for bespoke therapies, they summarize negative feedback on existing regulations from a variety of stakeholders and express their agreement. The authors remark that additional suggestions to the framework are welcome, which may indicate that a draft guidance and/or town hall meeting are on the horizon.
It certainly appears that the FDA aims to partner with therapeutic developers to get personalized medicines over common regulatory hurdles, and that they hope to keep pace with scientific innovation, yet it remains unclear exactly how the pathway will be implemented. Once official guidelines are published, the plausible mechanism pathway could theoretically provide much-needed clarity for developers of complex biologics, helping accelerate clinical development timelines and getting these life-changing therapies to patients faster.