A Comprehensive How-To Guide
Nucleic acid detection technologies form the backbone of modern molecular diagnostics, agricultural testing, and environmental monitoring. While PCR and qPCR remain industry standards, their reliance on thermal cycling and complex instrumentation limits their utility in rapid, field-deployable, or point-of-care settings.
To address these limitations, isothermal amplification methods combined with CRISPR-based detection have emerged as powerful alternatives. Recombinase Polymerase Amplification (RPA), when paired with sequence-specific CRISPR nucleases, enables rapid amplification and highly specific detection within a single isothermal workflow.
This guide outlines the mechanistic basis of RPA–CRISPR synergy and provides practical guidance for assay design, implementation, and optimization.
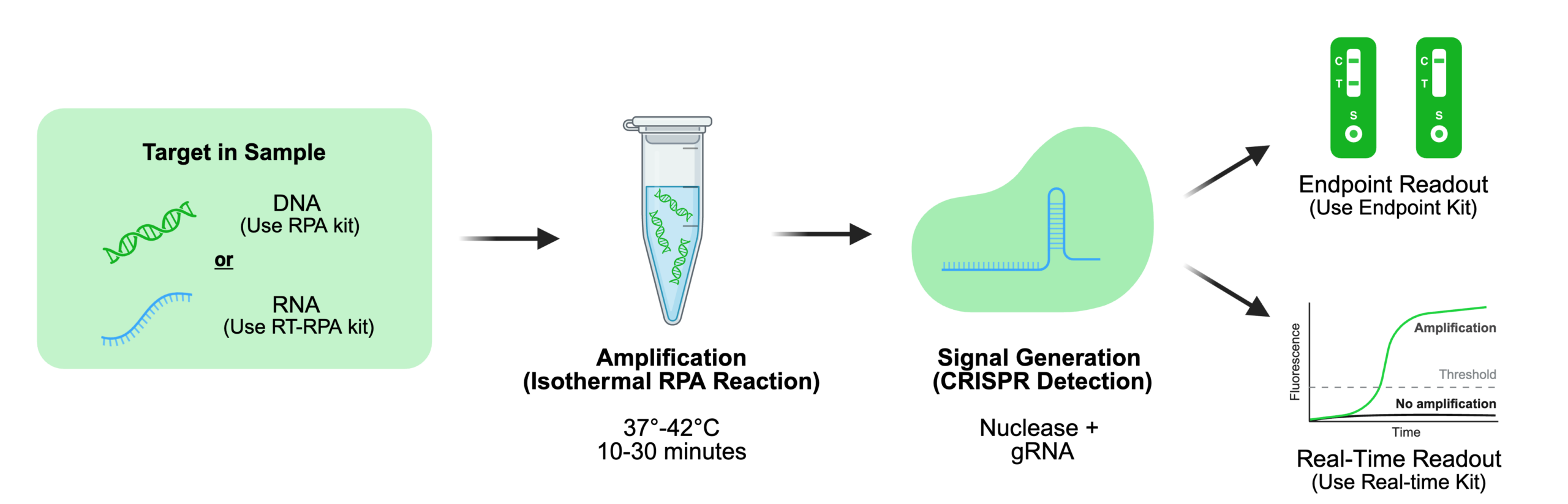
RPA and CRISPR address complementary limitations within an integrated workflow: RPA rapidly amplifies low-abundance nucleic acids but can generate non-specific products and is generally less discriminating at single-nucleotide resolution, making single-nucleotide polymorphism (SNP) detection particularly dependent on careful primer design and optimization. In contrast, CRISPR systems provide high sequence specificity through guide-directed recognition but require sufficient target input for robust activity. Together, RPA functions as the amplification engine and CRISPR as the specificity filter, enabling rapid, sensitive, and highly specific detection. To support this synergy, we provide a modular platform spanning RPA chemistries (endpoint, real-time, and RT-RPA), CRISPR nucleases (hfCas12Max and Cas9 variants), and synthetic guide RNAs—enabling rapid iteration and reproducible assay development across diverse assay architectures.
Core Mechanism: Recombinase Polymerase Amplification (RPA) enables rapid nucleic acid amplification at a constant temperature (37–42°C). The reaction is driven by recombinase–primer complexes, single-stranded DNA binding proteins (SSBs), and a strand-displacing polymerase. Recombinases facilitate primer invasion of homologous sequences within double-stranded DNA without thermal denaturation. The displaced strand is stabilized by SSBs, allowing polymerase extension and continuous amplification. Learn more in our RPA and RT-RPA Mechanisms and Methods Guide.
Our RPA kits (endpoint, real-time, and RT-enabled formats) provide optimized reagent systems for rapid isothermal amplification across DNA and RNA-derived templates. These kits are fully customizable, including lyophilized and lyo-ready formats, tailored buffer compositions, and configurations designed for assay-specific workflows.

The success of an RPA–CRISPR diagnostic tool depends largely on the specific properties of the effector protein used. While RPA provides sensitivity by rapidly copying the target DNA at a constant temperature, the guide RNA-nuclease complex determines how accurately the test identifies the target and how it triggers a readable signal. Selecting the right enzyme family requires matching the target (DNA or RNA) with the enzyme's ability to perform collateral cleavage—the "side-cutting" activity required to release a fluorescent signal for detection.
The following table provides a comparative breakdown of the most common nuclease families, highlighting their requirements and operational tradeoffs.
| Nuclease Family | Primary Target | Collateral Activity | PAM / PFS | Key Characteristics |
|---|---|---|---|---|
| Cas12 Family | dsDNA | Yes (ssDNA) | PAM Required |
Pro: Robust signal for DNA assays. Con: PAM limits targetable sites. |
| Cas13 Family | ssRNA | Yes (ssRNA) | Varies; some variants have PFS preferences. |
Pro: RNA sensing enabled; high turnover. Con: RNA stability/buffer issues. |
| Cas14 (Cas12f) | ssDNA | Yes (ssDNA) | No PAM for ssDNA; PAM required for dsDNA. |
Pro: Ultra-compact; great for SNPs. Con: Weaker on dsDNA targets. |
| Cas9 | dsDNA | No | PAM Required |
Pro: Highly flexible gRNA design. Con: Lacks collateral signal amplification. |
| Engineered Variations | DNA or RNA | Tuned | Varies |
Pro: High specificity; low off-targets. Con: Requires precise temp/buffer tuning. |
To this date, Cas12 and Cas13 systems are the primary drivers of signal generation in CRISPR-based assays due to their collateral cleavage activity. Cas12 is most commonly used for DNA detection following RPA amplification, while Cas13 enables RNA detection in both direct and amplification-coupled workflows. For DNA-targeted detection workflows, engineered Cas12 nucleases are often explored to improve signal-to-noise performance. Synthego’s hfCas12Max, a high-fidelity Cas12i-family nuclease, is designed to reduce non-specific collateral activity while retaining target-triggered activation and can be evaluated in CRISPR-based detection workflows.
Cas12 and Cas13 systems convert target recognition into measurable outputs through fluorescent reporter cleavage. Fluorescent reporters support real-time or quantitative detection, while lateral flow formats enable rapid, instrument-free readouts. In lateral flow systems, signal interpretation depends on reporter cleavage state, where intact and cleaved reporters produce distinct visual outputs. The choice between endpoint and real-time detection ultimately depends on assay requirements such as sensitivity, quantitation, and available instrumentation, with each format offering trade-offs in speed, complexity, and data resolution. For a deeper comparison, see our endpoint versus real-time RPA guide.
In contrast, Cas9 lacks collateral activity and is typically used in engineered detection systems where signal generation is externally coupled. While this makes Synthego enzymes like SpCas9 and high-fidelity variants such as eSpOT-ON exceptionally precise and reliable for applications like gene editing—where controlled, site-specific cleavage is essential—it limits their utility in diagnostics. Without intrinsic signal amplification through collateral cleavage, Cas9-based assays often require additional components or steps, increasing complexity and potentially reducing speed and sensitivity compared to systems built around enzymes like Cas12 or Cas13.
CRISPR targeting is constrained by sequence requirements: Cas12 relies on protospacer adjacent motifs (PAMs), while Cas13 does not require a PAM but may exhibit protospacer flanking site (PFS) preferences that influence targeting efficiency. Recent advances have introduced PAM-independent detection strategies—such as engineered primer ratios (e.g., asymmetric RPA) that generate ssDNA substrates—enabling Cas12-mediated detection without reliance on canonical PAM sequences and significantly expanding the range of targetable regions. Complementary approaches that pair these PAM-free workflows with iterative crRNA design further enhance assay specificity and robustness, particularly for challenging targets.
The precision of any CRISPR-based diagnostic is entirely dependent on the design of the guide RNA (gRNA), which serves as the search engine for the Cas enzyme. While the RPA step provides the bulk of the material, the gRNA ensures that only the correct sequence triggers a signal. A primary concern in design is off-target activity, where the gRNA might bind to a sequence with 1–3 mismatches, potentially causing false positives. To mitigate this, computational tools are used to screen gRNAs against the host genome and related pathogens, ensuring the "spacer" sequence is unique to the target. Synthego single guide RNAs (sgRNAs) provide a consistent and scalable source of sequence-specific targeting, enabling rapid assay optimization and streamlined workflow integration across CRISPR-based detection systems.
Furthermore, when multiplexing, gRNAs must be meticulously screened for cross-reactivity. In these high-plex scenarios, the challenge is ensuring that the gRNA for Target A does not inadvertently activate the Cas protein in the presence of Target B, while also maintaining a stable secondary structure that prevents the gRNAs from binding to one another rather than the viral template.
Developing a RPA-CRISPR diagnostic is best approached as a modular, stepwise process. Instead of optimizing everything at once, the workflow separates amplification (RPA) and detection (CRISPR) into independent systems. Each is validated on its own before being integrated into a single assay. This separation is important: most assay failures are difficult to diagnose because it's unclear if the problem lies in amplification, detection, or their integration. A modular workflow removes this guesswork by isolating each stage.
Before beginning assay design, it is essential to define both the starting material and the intended amplification format, as these parameters dictate the appropriate chemistry and kit selection.
Note: The "amplification readout" refers specifically to signal monitoring associated with the RPA reaction itself. This is independent of the final diagnostic readout generated by downstream detection systems—such as CRISPR-Cas cleavage, lateral flow strips, or fluorescent reporters—in either one-pot or two-pot workflows.
Establishing these parameters upfront ensures the selection of the correct enzymatic system and helps prevent workflow mismatches between the amplification and downstream detection stages.

The ideal amplicon is typically 80–200 bp. This range balances fast amplification with accessibility for CRISPR detection. Shorter regions tend to amplify quickly but may reduce flexibility in guide design, while longer regions increase the risk of secondary structure that can interfere with detection.
At this stage, it is also essential to confirm:
Once the target region is validated, it becomes the foundation for both primer and guide RNA design.
With the target defined, the next step is to build the amplification system. RPA primers are designed first—typically starting with 3–5 primer pairs as candidates, each around 30–35 nucleotides. These are screened experimentally to identify the most efficient and specific pair. Importantly, RPA is tested on its own, without CRISPR components. This isolates amplification performance from downstream detection.
A successful RPA reaction should:
At the end of this step, you should have a single optimized primer pair that reliably generates clean amplicons.
In parallel with RPA optimization, the CRISPR detection system is developed independently. Multiple guide RNAs (typically 3–6 gRNAs) are designed within the validated amplicon region. These are screened using a synthetic target under fixed reaction conditions. Again, the system is tested in isolation using a defined DNA or RNA target, Cas enzyme, and reporter.
A strong CRISPR candidate will show:
At this point, the assay has two independently validated components: one for amplification and one for detection.
Once both modules are functional, they are combined for the first time using a two-pot system.
In this setup:
This dilution step is important because it reduces carryover of RPA reagents that might inhibit Cas activity. In most cases, no additional cleanup is required. However, if the synthetic control works but the RPA product does not, this often indicates reaction inhibition. In those cases, a quick bead or column cleanup can confirm whether the issue is chemical interference rather than assay design. At this stage, different primer pairs and gRNAs are systematically combined to identify the best-performing pair.
With a working RPA–CRISPR pair established, the system is now optimized as a single biochemical unit. Unlike earlier steps, changes here affect both amplification and detection simultaneously.
Key parameters include:
The goal is not just activity, but consistent, reproducible signal generation across runs.
Once the system is stable, it is adapted for deployment. At this stage, the key decision is whether to use a two-pot or one-pot format. The two-pot format is typically used during development because it allows independent optimization of RPA and CRISPR conditions. It offers maximum flexibility and sensitivity but increases handling steps and contamination risk. The one-pot format combines everything into a single reaction. This reduces user steps and contamination risk but requires careful tuning of buffer compatibility and enzyme stability. It is generally reserved for finalized assays.
The final assay format depends on the intended use case:
Each readout imposes different constraints on reaction chemistry and reporter design.
Before deployment, the assay must be validated beyond ideal laboratory conditions.
This includes:
These tests ensure the assay is robust to biological variability and not just optimized for synthetic targets.

For both DNA and RNA targets, the integration of In Vitro Transcription (IVT) is a critical component of the Cas13 detection workflow. Because Cas13 is an RNA-guided RNase, it cannot natively recognize double-stranded DNA amplicons. Consequently, even when starting with an RNA target, the template is first converted and amplified into DNA via RT-RPA to achieve exponential gain, necessitating a "re-conversion" back into RNA for CRISPR activation. The process hinges engineering a T7 promoter sequence as a tail on one of the isothermal amplification primers. During the initial amplification phase (RPA for DNA or RT-RPA for RNA), the T7 promoter is integrated into the resulting dsDNA amplicons.
While adding the IVT step increases the workflow complexity—typically requiring a two-pot process—the trade-off is a massive gain in sensitivity. This is achieved through a three-tier amplification cascade:
In platforms like SHERLOCK, the IVT and Cas13 steps are combined into a single reaction mix. This allows for real-time detection: as the T7 polymerase produces RNA, Cas13 immediately recognizes it and triggers a signal. This synergy ensures that even when starting with a few copies of DNA or RNA, the system produces a robust, high-confidence result, often reaching attomolar sensitivity.
Expanding CRISPR-based assays to detect multiple targets requires maintaining signal orthogonality while preventing competition between reactions occurring in the same mixture. In practice, multiplexing is achieved through a combination of detection design strategies and careful kinetic balancing during amplification
A widely used approach for in-tube multiplexing is the use of orthogonal CRISPR-associated enzymes with distinct target preferences and reporter requirements. For example, Cas12 enzymes are typically used for DNA targets and cleave single-stranded DNA reporters, while Cas13 enzymes are used for RNA targets and cleave single-stranded RNA reporters. When combined in a single reaction, this enables simultaneous detection of different target classes. To distinguish signals, each enzyme–reporter pair is assigned a unique fluorescent label (e.g., FAM for Cas12 and HEX for Cas13), allowing independent readouts with minimal spectral overlap.
For multiplexing within the same target class (e.g., multiple DNA or multiple RNA targets), assays generally rely on orthogonal guide RNAs paired with uniquely labeled or barcoded reporters, rather than differences in intrinsic cleavage behavior. This allows multiple targets to be resolved even when using a single effector protein. Because Cas12 and Cas13 also differ in their sequence recognition requirements (e.g., PAM constraints for Cas12 and PFS preferences for Cas13), they can be independently programmed to interrogate distinct targets within the same sample without cross-reactivity.
When instrumentation is limited or a visual output is preferred, multiplexing can be achieved through spatial separation of detection signals using lateral flow assays (LFAs).
In this format, amplification and CRISPR detection occur in a single reaction, but target identification is resolved downstream at the level of the test strip. Distinct capture reagents immobilized at separate test lines direct reporter accumulation to defined spatial positions, enabling each target to be read independently (e.g., Line 1 for Target A, Line 2 for Target B). This approach is particularly useful for field-deployable diagnostics, where binary or semi-quantitative interpretation is preferred over fluorescence-based readouts.
Regardless of detection strategy, the primary constraint in multiplex CRISPR assays is often upstream amplification rather than detection chemistry itself. When multiple primer sets are present in a single reaction (e.g., RPA or RT-RPA), competition for reagents can lead to uneven amplification efficiencies across targets.
To mitigate this, primer concentrations are frequently balanced to reduce amplification bias, particularly for fast-amplifying targets that might otherwise dominate reaction resources. In addition, primer design is critical to minimize dimer formation and nonspecific interactions, which can significantly reduce assay sensitivity in multiplex formats.
| Method | Mechanism | Best For |
|---|---|---|
| Orthogonal CRISPR systems | Multiple Cas enzymes or guide RNAs with distinct reporters | Fluorescence-based, high-sensitivity laboratory assays |
| Spatial separation (LFA) | Physical separation of capture lines on a strip | Field-deployable, instrument-free diagnostics |
RPA–CRISPR systems combine rapid isothermal amplification with highly specific nucleic acid detection, enabling sensitive and flexible diagnostic workflows without complex instrumentation.
With continued advances in engineered nucleases, optimized reaction systems, and modular reagent platforms such as those provided by Synthego, these technologies are increasingly positioned for broad adoption across research, translational, and applied diagnostic settings.
Visit our RPA Resource Hub for additional RPA materials.
In most optimized "two-pot" protocols, a cleanup step is not required. CRISPR enzymes (like Cas12 and Cas13) are remarkably robust and remain active in the presence of RPA reagents, including the high concentrations of PEG and proteins used in the isothermal mix. This allows users to simply transfer a small volume of the RPA product directly into the CRISPR master mix, which is essential for rapid, field-based testing.
The choice depends primarily on your target and preferred workflow:
Most Cas enzymes require a "handle" next to the target sequence to bind and activate.
When designing your RPA primers, you must ensure the amplified region contains these specific motifs, or the CRISPR enzyme will fail to recognize the target even if the RPA step is successful.
Yes, “one-pot” reactions are possible and highly desirable for reducing contamination and simplifying workflows. However, they are technically challenging to optimize because RPA typically runs best at 37–42 °C, while several Cas enzymes exhibit peak activity at ~42–45 °C—several degrees higher than the optimal temperature range most RPA reactions are designed around. This creates a need to balance reaction conditions so that both amplification and detection remain efficient.
In addition, the chemical components of the buffers (especially Mg²⁺ concentrations) must be carefully tuned so that both the recombinase polymerase and the Cas enzyme can function simultaneously without inhibiting one another.
This "dual-recognition" system provides two layers of specificity. First, the RPA primers must correctly identify and amplify the target sequence. Second, the CRISPR gRNA must find its specific match within that amplified product to trigger the signal. This significantly reduces "false positives" caused by primer-dimers or non-specific amplification, as the Cas enzyme will only activate if the exact target sequence is present.